



Next: 7.1.13 Dust: dustscheme=dust_moment04_c2
Up: 7.1 Parameter file: rhd.par
Previous: 7.1.11 Tensor-viscosity control
Contents
Index
Subsections
7.1.12 Dust/molecules/hydrogen ionization: General
CO5BOLD can handle a number of additional density arrays. They can be used to describe
e.g. the mass density of dust distribution moments or number densities of molecules.
These species are properly advected with the gas density.
There is also already a small number of dust/molecule formation models available.
These models have to be improved in the future and the influence on the radiation
field (opacities, radiation pressure on dust) has to be taken into account.
7.1.12.1 character dustscheme
A scheme for dust or molecule formation and transport can be selected e.g. with
character dustscheme f=A80 b=80 n='Dust model' &
c0='none (default), nosource, dust_simple_01, co_component01_01' &
c1='dust_k3mon_01, dust_k3mon_02'
dust_k3mon_01
The following values are currently possible:
none, None: No handling of any dust/molecule density at all.
nosource: Skip source term step for dust/molecules entirely, but do the transport.
dust_simple_01: Simple and unrealistic 'dust' formation model (only for testing of
the numerics).
co_component01_01: Simple CO formation (from Matthias Steffen)
with one component only but realistic time scales.
dust_k3mon_01: Simple C-rich dust formation (routines from Susanne Höfner)
with one component only but realistic time scales.
dust_k3mon_02: Simple C-rich dust formation (routines from Susanne Höfner)
with two components for dust density and free carbon density.
dust_k3mon_03: Simple forsterite dust formation (based on routines from Susanne Höfner)
with two components for dust density and free "forsterite monomer" density.
dust_bins_01: Multi-size-bin forsterite dust formation
(based on Rossow's equations, see Rossow, 1978)
with one bin for the monomers and several bins for the different grain sizes.
dust_moment04_c2: C-rich dust chemistry, 4 moments (routines from Susanne Höfner).
chemreacnet: chemical reaction networks (routines from Sven Wedemeyer-Böhm and Inga Kamp).
hiontd: time-dependent hydrogen ionization (routines from Jorrit Leenaarts and Sven Wedemeyer-Böhm).
7.1.12.2 character dustreconstruction
While usually (i.e., when this parameter is not set) the reconstruction scheme
for the additional density fields is the same as for the normal hydrodynamics
quantities (see 7.1.8.4),
it can make sense to choose a strictly monotonic scheme
for these additional fields.
This scheme can be more (e.g. PP)
or less (e.g. PPmimo) aggressive.
It can be chosen e.g. with
character dustreconstruction f=A80 b=80 n='Reconstruction method for qucs' &
c0=Constant c1=Minmod/VanLeer/Superbee c2=PP/PPmimo &
c3=FRmimo,FRmono
FRmono
Useful combinations are
reconstruction=vanLeer, dustreconstruction=Superbee.
reconstruction=FRweno, dustreconstruction=FRmono.
reconstruction=FRweno, dustreconstruction=PP.
reconstruction=HBweno, dustreconstruction=PP.
reconstruction=HBweno, dustreconstruction=PPmimo.
There are ten parameters (real c_dust01 to c_dust10)
to control each dust formation scheme in detail. A parameter can be given as in
real c_dust01 f=E15.8 b=4 n='Dust parameter 1'
0.0
The meaning (and unit) can vary from scheme to scheme.
The default value is 0.0 in each case.
Important:
The parameter real c_dust01 must be set to 1.0 in order to
activate advection of particle densities for the CHEM (chemreacnet) and the
HION (hiontd) module. The default value is 0.0, i.e. advection is switched off.
The name of the input file containing the chemical reaction network.
character chem_reacfile f=A80 b=80 n='file name of reaction table'
chem.dat
The path of the input file containing the chemical reaction network.
character chem_reacpath f=A80 b=80 n='path of reaction table'
/data/sven/cobold/dat/chem/
Abundance of the representative metal ('M', if present) relative
to hydrogen (
![$\epsilon_\mathrm{M} = [M] / [H]$](img215.png) ). A value of
). A value of 1.0E-04 means there
are 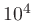 hydrogen atoms for every metal atom. The metal number density
for each grid cell is then derived via
hydrogen atoms for every metal atom. The metal number density
for each grid cell is then derived via
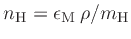 (assuming a pure
hydrogen gas).
This parameter is ignored when a
(assuming a pure
hydrogen gas).
This parameter is ignored when a quc array for the metal is found in the
input model.
real chem_abumetal f=E15.8 b=4 n='Chemical abundance of repres. metal'
1.0E-04
The path of all input files for HION.
character hion_datapath f=A80 b=80 n='HION data path'
/data/sven/cobold/dat/hion/
The file name of the model atom for HION.
character hion_atomfile f=A80 b=80 n='HION atom file name'
H_6.atom
The name of the HION input file containing the chemical abundances.
character hion_abufile f=A80 b=80 n='HION abundance file name'
abundance.input
The name of the HION input file containing the electron density table.
character hion_edensfile f=A80 b=80 n='HION electron density file name'
edens.dat
The name of the HION input file containing the partition functions.
character hion_pffile f=A80 b=80 n='HION partition function file name'
pf_kurucz.dat
Time increment for additional HION output. Positive values specify
the time increment in seconds, negative values the increment in
computational time steps. Setting the parameter to zero, suppresses
the output.
real dtime_out_hion f=E15.8 b=4 n='Output file time step (HION)'
-10.0
Number of chunks to use for HION in order to limit the required memory.
Do not make it bigger than the number of points in the x2 direction.
integer hion_chunks f=I9 b=4 n='Number of HION chunks'
1
Next: 7.1.13 Dust: dustscheme=dust_moment04_c2
Up: 7.1 Parameter file: rhd.par
Previous: 7.1.11 Tensor-viscosity control
Contents
Index